زیست شناسی
این وبلاگ برای تمام عزیزانی که به علوم زیستی علاقه دارند ایجاد شده و از همه چیز و همه جا که مرتبط با زیست شناسی باشد اطلاعات میدهدزیست شناسی
این وبلاگ برای تمام عزیزانی که به علوم زیستی علاقه دارند ایجاد شده و از همه چیز و همه جا که مرتبط با زیست شناسی باشد اطلاعات میدهددرباره من
 ***دانش آموخته دانشگاه تهران -
**** ***فوق لیسانس فیزیولوژی جانوری***
-دبیر زیست شناسی دبیرستان ماندگار البرز
ادامه...
***دانش آموخته دانشگاه تهران -
**** ***فوق لیسانس فیزیولوژی جانوری***
-دبیر زیست شناسی دبیرستان ماندگار البرز
ادامه...
با بیماری پلی سیتمی آشنا شویم
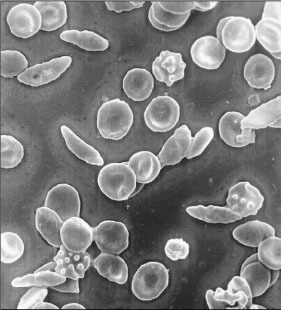
پلی سیتمی عکس آنمی
شرح بیماری
پلی سیتمی افزایش تعداد گلبولهای قرمز خون. این اختلال بیشتر در بزرگسالان بالای 50 سال بروز میکند (ولی محدوده سنی آن 90-15 سال است) و در آقایان شایعتر است. این بیماری سه نوع دارد:
- پلی سیستمی ثانویه (پلی سیستمی واکنشی)، که عارضهای از بیماری یا عواملی غیر از اختلالات سلولهای خون است.
- پلی سیستمی استرسی
- پلی سیستمی کاذب که به کاهش حجم پلاسمای خون مربوط میباشد.
علایم شایع
برخی بیماران هیچ علامتی ندارند. سایر بیماران ممکن است دارای هر یک از علایم زیر باشند:- خستگی؛ سردرد؛ خواب آلودگی؛ منگی
- خارش یا پرخونی پوست
- بزرگی طحال
- خونریزی بدون توجیه
علل
پلیسیستمی حقیقی: ناشناخته
پلی سیستمی ثانویه: بیماریهای مادرزادی قلب، بیماریهای مزمن ریه، کشیدن سیگار معمولی یا سیگار برگ، زندگی در ارتفاعات بالا
پلیسیستمی استرس: مصرف داروهای ادرارآور، استعمال دخانیات، کم آبی بدن
عوامل افزایش دهنده خطر
۱-استعمال دخانیات
۲-بیماریهای قلبی یا ریوی
۳-استرس
۴-سابقه خانوادگی پلیسیستمی
پیشگیری
- پلیسیستمی حقیقی در حال حاضر قابل پیشگیری نیست.
- پیشگیری از پلیسیستمی ثانویه یا پلیسیستمی استرس: خودداری از استعمال دخانیات جلوگیری از کم آبی بدن اقدام به درمان طبی بیماریهای قلبی یا ریوی
عواقب مورد انتظار
- پلیسیستمی حقیقی غیر قابل علاج است ولی علایم آن با درمان قابل کنترل است. متوسط مدت زنده ماندن بیماران با درمان 15-7 سال است؛ حتی برخی بیماران ممکن است 20 سال یا بیشتر نیز زنده بمانند.
- سایر انواع پلیسیستمی با برطرف کردن علت زمینهای قابل درمانند.
عوارض احتمالی
- تشکیل لخته درون وریدها یا شریان ها
- نقرس
- سکته مغزی
- حمله قلبی
- زخم گوارش
- سنگ کلیه
- لوسمی(نوعی سرطان خون)
درمان
اصول کلی
- بررسیهای تشخیصی ممکن است شامل بررسیهای آزمایشگاهی مغز استخوان و خون (شمارش گلبولهای قرمز خون، اندازهگیری هماتوکریت)، اشعهنگاری کلیه ها و بررسی گلبولهای قرمز با کروم رادیواکتیو باشد.
- مراحل درمانی بر اساس وضعیت هر بیمار متفاوت بوده و به سن بیمار، مدت بیماری، نوع پلیسیتمی، عوارض، و فعالیت در زمان ابتلا به این بیماری بیماری بستگی دارد.
- مراحل درمانی احتمالی برای حفظ هماتوکریت در نزدیک محدوده طبیعی و جلوگیری از تشکیل لخته یا خونریزی عبارتند از: فصد (گرفتن خون اضافی بدن از طریق ورید)؛ درمان رادیو ایزوتوپ؛ و دارو درمانی. درمان انتخاب شده بستگی به علایم و پاسخ به درمان دارد. در مواردی ممکن است به بیش از یک نوع درمان نیاز باشد.
داروها
- آسپیرین برای جلوگیری از لخته شدن خون و کاهش احتمالی سکته مغزی یا حمله قلبی ممکن است توصیه گردد.
- فسفر رادیواکتیو سیتوتوکسیک
- آلوپورینول برای کاهش دادن سطح بالای اسید اوریک
- داروهای ضد خارش
- داروهای مهار کننده گرنده H2 یا داروهای ضد اسید برای مهار تولید بیش از حد اسید معده
فعالیت در زمان ابتلا به این بیماری
پس از درمان، فعالیت در زمان ابتلا به این بیماری های طبیعی خود را هر چه سریعتر از سر بگیرید.
رژیم غذایی
رژیم خاصی نیازی نیست. برای حفظ مایعات بدن در حد مطلوب هر دو ساعت 250 سیسی مایعات بنوشید.
درچه شرایطی باید به پزشک مراجعه نمود؟
- اگر شما یا یکی از اعضای خانوادهتان دارای علایم پلیسیستمی باشید.
- بروز علایمی که نشانه عوارض پلیسیستمی هستند.
- اگر دچار علایم جدید و غیرقابل توجیه شده اید. داروهای تجویزی ممکن است با عوارض جانبی همراه باشند.
منبع: www.ireb.com
با بیماری ارثی فنیل کتونوریا بیشتر آشنا شوید
فنیل کتونوریا

یک بیماری ژنتیکی متابولیکی است که به علت کمبود یا فقدان آنزیم فنیل آلانین هیدروکسیلاز و تبدیل فنیل آلانین ( یک اسید آمینه ضروری ) به متابولیت هایی که تظاهرات اصلی آن عقب ماندگی شدید ذهنی ، پرفعالیتی همراه با تحرکات بی هدف و تشنج است. اغلب بیماران به حدی دچار عقب ماندگی می شوند که نیاز به مراقبت 24 ساعته یا نگهداری تمام وقت در آسایشگاه دارند.
این بیماری ژنتیکی از نوع اتوزومال مغلوب است و الگوی ارثی آن تحت ازدواج فامیلی قرار دارد و تا 50 درصد شانس بروز را افزایش می دهد. بروز بیماری در ایران 1 در 6000 تا 8000 تولد است. در کشورهای اروپایی همجوار یا نسبتاً همجوار ما بروز بین 1 در 4000 تا 10000 تولد می باشد.
متوسط سن تشخیص این بیماری 2 سالگی است که در این سن صدمات مغزی کامل شده و غیرقابل برگشت است. کودک مبتلا در ابتدای تولد طبیعی است ولی به تدریج عقب ماندگی ذهنی پیشرفت می کند و طی چند ماه آشکار می شود. اگر کودک تحت درمان قرار نگیرد به ازاء هر یک ماه 4 نمره از IQ وی کاسته می شود و تا پایان سال اول 50 نمره از IQ را از دست خواهد داد.
با توجه به اینکه پیشگیری از بروز پیشگیری از بروز عقب ماندگی ذهنی در این بیماری بسیار آسان است. لذا در آزمایشات غربالگری بدو تولد گنجانده شده است و برای کلیه نوزادان انجام می گیرد تا در صورت تائید، اقدامات درمانی و رژیم غذایی اعمال شود.
برگرفته از : دانشگاه علوم پزشگی فارس
سندرم کلاین فتتر
سندرم کلاین فلتر
سندروم کلاین فیلترطیفی ازاشکال فنوتیپی است که دراثرافزایش تعداد کروموزوم X به دو عدد
یا بیشتر در کنار وجود یک یا چند کروموزوم Yایجاد می شود.
شیوع سندروم کلاین فیلتر47 XXY یک در هزار مذکر زنده متولد شده است.
وجود ان در سقط های خودبه خودی بسیار نادر بوده و کاریوتایپ های دیگر به غیر 47 xxy
نیز نادر است.اشکال سوماتیکی این سندروم بسیار کم و غیر اختصاصی است.
این بیماری علی القاده قبل از بلوغ تشخیص داده نمی شود مگر اینکه در برنامه ی بیمار یابی
بعنوان مثال در مؤسسات عقب ماندگی ذهنی به کروماتین x برخوردکرده باشیم وجود کوچکی
انگشتان با کوتاهی خط بر جسته به تشخیص کمک می کند.
سندروم کلاین فیلتر با بالا بودن سن مادر ارتباط دارد.این مسًله یک مشکل جدی اطفال نیست
و به غیر از مشکل نازایی اغلب مردان مبتلا زندگی طبیعی دارند.
سندرم ترنر

سندرم ترنر
سندرم ترنر چیست؟
سندرم ترنر یک ناهنجاری کروموزومی است که در آن نوزاد دختر، به جای داشتن دو کروموزوم جنسی X، تنها با یک کروموزوم X یا دو کروموزوم که یکی آسیب دیده، متولد میشود. در بیشتر موارد زنان مبتلا به این ناهنجاری، در صورت درمان نشدن، بسیار کوتاه قد (حداکثر 140 سانتی متر) بوده و به مشکلات فیزیکی و سلامتی متعددی نیز دچار هستند.
از آنجایی که زنان مبتلا به سندرم ترنر فاقد سیستم تخمدانی سالم هستند، معمولا در سن بلوغ خصوصیات ثانویه زنانه به طور کامل در آنها به وجود نمی آید و آنها نازا هستند. البته پیشرفتهای علم پزشکی از جمله در هورمون درمانی و باروری خارج از رحم، میتواند به این زنان کمک کند.
از مشکلات دیگر این سندرم، داشتن ناهنجاری کلیه ها و قلب، ابتلا به بیماریهایی چون فشار خون بالا، چاقی، دیابت، آب مروارید، مشکلات غده تیرویید و آرتروز است. دختران مبتلا به سندرم ترنر معمولا از هوش عادی برخوردارند اما در آنها مواردی از کند ذهنی و اشکال در یادگیری به خصوص در ریاضیات، دیده شده است.
مشکلات شنوایی نیز در دختران مبتلا به سندرم ترنر بیشتر است.هرچند در این موارد آمار فزاینده ای از مشکلات روحی وجود ندارد،اما طبیعی است که بسیاری از این دختران دچار مشکلاتی چون فقدان اعتماد به نفس باشند. کودکان مبتلا به این ناهنجاری معمولا بیش فعالند. علی رغم تفاوتهای جسمی و مشکلات احتمالی ناشی از TS، این افراد با کمک درمان سریع و حمایت دائمی خانواده خود میتوانند زندگی طبیعی، سلامت و پرباری داشته باشند.

چگونگی تشخیص سندرم ترنر
یک پزشک با کمک معاینه و مشاهده نشانه های زیر میتواند وجود سندرم ترنر را تشخیص دهد:
• قد کوتاه
• وجود بخشی پرده مانند در قسمت پوست بین گردن و شانه
• خط رویش موی پایین در پشت سر
• شکل غیر عادی چشمها از جمله افتادگی پلک
• ناهنجاری رشد استخوانها به خصوص در دست و آرنج
• عدم شکل گیری سینه ها در سن بلوغ
• عدم بروز قاعدگی
• وجود بیش از حد خال بر روی پوست
بروز علائم و خصوصیات سندرم ترنر در افراد مختلف بسیار متفاوت است.عده ای دارای بسیاری از خصوصیات آن بوده و عده دیگر تنها تعداد اندکی از آنها را دارا هستند.
یک تست خون به خصوص که در آن به بررسی کروموزومهای فرد پرداخته میشود و کاریوتایپ (karyotype) نام دارد، میتواند به پزشکان کمک کند تا به سرعت متوجه TS شده و به کنترل آن بپردازند. تعداد کروموزومها در تست کاریوتایپ مارش میشود و افراد مبتلا به سندرم ترنر، به جای 46 عدد، دارای 45 کروموزوم هستند.
کنترل سندرم ترنر
از آنجایی که این ناهنجاری کروموزومی است، هیچ درمانی برای آن وجود ندارد اما درحال حاضر روشهایی برای بهبود بخشیدن به شرایط این افراد موجود است.
• درمان با هورمون رشد به تنهایی یا با هورمونهای دیگر، اگر به موقع انجام شود، میتواند به میزان رشد افزوده و حداکثر قد در دوران بلوغ را افزایش دهد.
• درمان با جایگزینی استروژن معمولا در سنین 12 یا 13 سال آغاز میشود و میتواند در بروز خصوصیات ثانویه جنسی موثر باشد.اما نمیتواند موجب درمان نازایی شود.
• جراحی قلب در مورد مواردی که به ناهنجاری قلب مبتلا هستند، لازم است و نتایج مطلوبی دارد.
• دانش امروز به زنان مبتلا به TS کمک میکند که باردار شوند. به این ترتیب که یک تخمک اهدایی بارور شده را پرورش داده و رویان را در رحم شخص مورد نظر جاسازی میکنند. به کمک هورمون درمانی، این زن میتواند جنین آزمایشگاهی را حفظ نموده و زایمان کند.
تفاوتها را بشناسیم
بسیاری از دختران مبتلا به سندرم ترنر از هوش طبیعی برخوردارند و آن دسته اندکی که دارای ناتوانیهای یادگیری هستند میتوانند در مراکز ویژه به پیشرفتهای چشمگیری دست یابند. پس نباید با فرزندان یا اطرافیان مبتلا به TS مانند یک فرد کم هوش یا ناتوان برخورد نماییم. این زنان ظریف و کوچک اندام، میتوانند افرادی بسیار تاثیر گذار و موفق باشند و باید بدانند که علی رغم مشکلاتی که سلامت جسمیشان را تهدید میکند، میتوانند به فعالیتهای گوناگون و مورد علاقه خود پرداخته و یک فرد عادی باشند.
منبع : مجله اینترنتی فریا
سندرم داون

سندرم داون یا منگولیسم
اگرچه سندرم داون قابل پیش گیری نیست، اما میتوان قبل از تولد کودک آنرا تشخیص داد. مشکلات جسمی و سلامتی همراه با دی.اس. قابل درمان هستند، و در هر جامعه امکانات و منابع مختلفی برای کمک به این کودکان و والدین آنها وجود دارد.
علت بروز سندرم داون چیست؟
به طور معمول، هر کودک در زمان لقاح اطلاعات ژنتیکی خود را به وسیله 46 کروموزوم از والدین خود به ارث میبرد: 23 کروموزوم از مادر و 23 کروموزوم از پدر. اما در اکثر موارد بروز سندرم داون، کودک یک کروموزوم اضافه دریافت میکند و به جای 46، 47 کروموزوم به او منتقل میشود. این ماده اضافه ژنتیکی موجب تاخیر در رشد جسمانی و عقلانی کودک میشود.
هر چند کسی به طور قطع نمیداند که چرا این حالت اتفاق می افتد و هیچ روشی برای پیش گیری از اشتباه کروموزومی که موجب این نارسایی میشود، شناخته نشده است. تا مدتها تصور میشد بیشتر مبتلایان به دی.اس. از مادران بالای 35 سال به وجود آمده اند، اما آمار وتحقیقات پزشکی جدید ثابت کرده است که 80 درصد کودکان مبتلا به سندرم داون از زنانی زیر 35 سال متولد شده اند، البته بالا رفتن سن مادر شانس ابتلا به این سندرم را افزایش میدهد اما مهمترین دلیل بروز آن نیست.
کودکان مبتلا به سندرم داون دارای خصوصیات ظاهری مشابه هستند که از جمله بارزترین آنها میتوان به نیمرخ مسطح، چشمان مورب رو به بالا، گوشهای کوچک، یک تک خط در وسط کف دست و زبان بزرگ اشاره کرد. یک پزشک معمولا با یک معاینه جسمی میتواند بگوید که یک نوزاد دچار چنین نارسایی هست یا خیر.
معمولا این کودکان در زمان تولد قد و وزن معمولی دارند اما به طور کلی رشد آنها از سرعت کمی برخوردار است و از همسالان خود کوچکتر به نظر میرسند. در کودکان زیر 2 سال، کشیدگی کم و حرکات محدود ماهیچه موجب بروز اشکالاتی در مکیدن و غذا خوردن و همچنین مشکلات هاضمه دیگری چون یبوست میشود. کودکان نوپا و بزرگتر نیز ممکن است در حرف زدن، یادگیری و انجام کارهای شخصی چون غذا خوردن، لباس پوشیدن و دستشویی رفتن تاخیر زیادی داشته باشند.
سندروم داون به اشکال متفاوتی بر تواناییهای شناختی کودکان تاثیر میگذارد، اما بیشتر آنها دچار عقب افتادگی ذهنی خفیف تا میانه هستند. کودکان مبتلا به دی.اس. میتوانند بیاموزند و در طول زندگی خود قادر به پرورش مهارتهایی هستند. به زبان ساده، آنها با سرعتی متفاوت از دیگران به اهداف خود دست می یابند - و به همین دلیل بسیار مهم است که آنان را کودکان دیگر و حتا کودکان با شرایط مشابه، مقایسه نکنیم - .
این کودکان دارای طیف وسیعی از تواناییها هستند و هیچ راهی وجود ندارد تا در هنگام تولد پیش بینی شود که آنها در چه زمینه ای موفق خواهند بود.
مشکلات طبی مربوط به سندرم داون
در حالی که بعضی کودکان دارای دی.اس. هیچ مشکل خاص سلامتی ندارند، عده ای از آنها دچار انواع مشکلات طبی هستند که نیازمند توجه و رسیدگی خاص است. برای مثال، نیمی از تمام کودکانی که با سندرم داون متولد میشوند دچار نقص قلب مادرزادی بوده و مستعد ابتلا به فشارخون ریوی هستند. این مشکلات توسط کاردیوگرافی اطفال قابل شناسایی هستند و بیشتر آنها با دارو یا جراحی رفع میشوند.
تقریبا نیمی از این کودکان دارای مشکلات شنوایی و بینایی هستند. کاهش شنوایی میتواند به دلیل جمع شدن مایع در گوش میانی یا ناهنجاری ساختمان گوش باشد. مشکلات بینایی معمولا شامل تاربینی (تنبلی چشم)، نزدیک یا دور بینی و افزایش احتمال ابتلا به آب مروارید است. در مورد چنین کودکانی، مراجعه برای سنجش بینایی و شنوایی بسیار ضروری است تا قبل از اینکه هر مشکل احتمالی توانایی صحبت کردن یا مهارتهای دیگر آنها تحت الشعاع قرار دهد، در رفع آن بکوشند.
از دیگر نارساییها معمول در مبتلایان به سندرم داون میتوان به مشکلات غده تیروئید، ناهنجاری روده، مشکلات تنفسی، چاقی، حساسیت بیش از حد به عفونت و شانس فزاینده ابتلا به سرطان خون را نام برد. خوشبختانه بسیاری از این حالات قابل درمان هستند.
اگر شما فرزندی مبتلا به سندرم داون دارید، حتما در ابتدا غرق در احساس شکست، گناه و ترس بوده اید. گفتگو با والدین دیگری که چنین فرزندی دارند میتواند به شما کمک کند تا بر احساسات خود فائق شوید و راه هایی برای مقابله با مشکلات بیابید. عده ای از والدین اعتقاد دارند که هرچه بیشتر در باره این مشکل ژنتیکی بدانند، ترس کمتری از آینده خواهند داشت.
هنگامی که کودک شما به سن مدرسه رسید، بستگی به میزان درک و دریافت او و همچنین قابلیتهایی که دارد میتواند به مدرسه کودکان استثنایی و یا به مدرسه معمولی برود. مهم این است که به خاطر احساسات نادرست، کودک را از رفتن به مدرسه باز نداریم. با کارشناسان مشورت کنید و بهترین محل را برای تحصیل کودک خود انتخاب کنید.
امروزه بسیاری از کودکان مبتلا به سندرم داون به سن مدرسه میرسند و از بسیاری فعالیتهای مناسب سنشان بهره مند هستند. عده کمی از آنها به کالج هم رفته اند. بسیاری از آنها توانسته اند به یک زندگی نیمه مستقل دست یابند و عده دیگری که همچنان در خانه والدین خود زندگی میکنند در حدی هستند که میتوانند شغلی داشته باشند و در اجتماع به موفقیتهایی دست یابند.
منبع: مجله اینترنتی فریا
اندامک کلروپلاست
مقدمه
کلروپلاست معمولا از میتوکندری بزرگتر است و شباهت زیادی به میتوکندری دارد و جایگاه فرآیند فتوسنتز میباشد. کلروپلاستها جز گروهی از اندامکها هستند که این اندامکها پلاستید نام دارند. پلاستیدها در کلیه سلولهای گیاهی یافت میشوند و شامل اتیوپلاست ، کلروپلاست ، کروموپلاست ، آمیلوپلاست و الایوپلاست هستند.
وجه مشترک تمام پلاستیدها این است که تمام آنها از اندامک کوچک اولیهای به نام پروپلاستید ایجاد میشوند. پروپلاستید که پیش ساز کلیه پلاستیدها است. بسته به بافت گیاه و پیامهای محیطی به انواع گوناگون پلاستها تمایز پیدا میکند. کلروپلاست تنها پلاستیدی است که کلروفیل دارد و عمل فتوسنتز را انجام میدهد.
تاریخچه
کلروپلاستها به دلیل رنگ داشتن رنگ سبز از اولین اندامکهایی هستند که در یاختههای گیاهی نظر پژوهشگران را به خود جلب کردهاند. ووشر در سال 1803 رده بندی جلبکهای رشتهای آب شیرین را بر بنای شکل ذرات سبز موجود در آنها قرار داد و آنها را به کونفروهای مارپیچی ، ستارهای و لولهای تقسیم کرد. در جلبکها کلروپلاستها ساختمان سادهتری دارند و اغلب آنهارا کروماتوفور مینامند. در گیاهان پیشرفته و عدهای از جلبکهای سرخ و قهوهای کلروپلاستها کروی ، بیضوی و یا اغلب عدسی شکل هستند.
اندازه کلروپلاست
کلروپلاستها اندازه بسیار متفاوتی دارند. طول آنها از حدود 2 تا بیش از 30 میکرون میرسد. در گیاهان پیشرفته طول کلروپلاستها 3 تا 10 میکرومتر ، عرض آنها 1 تا 3 و ضخامتشان 1 تا 2 میکرومتر است. اندازه کلروپلاست به ویژگیهای وراثتی ، سن یاخته و دیگر ویژگیهای فیزیولوژیکی یاخته وابسته است. یاختههای پلی پلوئید کلروپلاستهای درشتتری از یاختههای دیپلوئید دارند.
رنگ کلروپلاست
کلروپلاستها به دلیل داشتن کلروفیل اغلب سبز رنگ هستند اما در برخی شرایط فیزیولوژیکی یا بر حسب نوع یاخته و میزان نسبی رنگیزههای غیر کلروفیلی ممکن است به رنگهای دیگری دیده شوند. در جلبکهای قهوهای و قرمز ، رنگ سبز کلروفیل بوسیله سایر رنگیزهها پوشیده شده است.
تعداد و محل کلروپلاست
تعداد کلروپلاست بر حسب نوع یاخته ، گونه گیاهی و سن یاخته تغییر میکند. تعداد کلروپلاستها در هر میلیمتر مربع برگ کرچک به حدود 400 هزار میرسد و یک درخت ممکن است تا 1012 عدد کلروپلاست داشته باشد. کلروپلاستها در یاختههای جلبکها و گیاهان مختلف در بخشهای مختلف یاخته قرار میگیرند. بطور معمول در بخشهای کناری یاخته که امکان دریافت نور بیشتر است فراوانی بیشتری دارند.

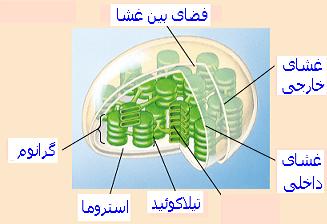
در ساختمان کلروپلاستها سه بخش اصلی شامل پوشش پلاستی ، ماده زمینهای یا استروما و ساختمانهای غشایی درونی قابل تشخیص است.
پوشش پلاستی
غشای خارجی
غشای خارجی کلروپلاست ضخامت متوسط حدود 60 آنگستروم دارد و از نوع غشاهای زیستی واحد است. این غشا صاف است، ریبوزوم ندارد و سد بین سیتوزول و درون پلاست است.
اتاق خارجی
اطاق خارجی یا فضای بین دو غشا وسعت متوسط حدود 100 تا 200 آنگستروم دارد و از مایعی دارای آب ، ترکیبات مختلف آلی ، مقدار کمی نمکهای کانی و یونهای حاصل از آنها پر شده است.
غشای داخلی
این غشا ویژگیهای عمومی شبیه غشای خارجی دارد. ضخامت متوسط آن حدود 60 آنگستروم است. گرچه غشای داخلی میتواند چین خوردگیهایی را به درون پلاست داشته باشد. اما نظریه کنونی بر این است که سیستمهای غشایی درونی کلروپلاست اساسا مستقل از غشای داخلی است.
اتاق داخلی
ماده زمینهای یا استروما اطاق داخلی کلروپلاست را پر کرده است. در استروما اجزای قابل رویت با میکروسکوپ الکترونی مانند سیستم غشاهای درونی ، مولکولهای DNA مشابه با پروکاریوتها ، ریبوزومهای از نوع 70s به حالت منفرد یا پلیزوم. در استروما اغلب ذرات نشاسته نیز وجود دارد. استروما دارای آنزیمهای مختلف از جمله آنزیمهای واکنشهای مرحله تاریکی فتوسنتز و آنزیمهای لازم برای بیوسنتز پروتئینهاست.
سیستم غشایی درون کلروپلاست
در استرومای کلروپلاستها ساختمانهای غشایی زیادی وجود دارند که مقدار آنها و نوع آرایششان به حسب نوع گیاه و ویژگیهای فیزیولوژیکی یاختهها متفاوت است. این ساختمانها تیلاکوئید نام دارند. این غشاها با سازمان یافتگی بسیار ویژه خود جایگاه انجام واکنشهای مرحله نوری فتوسنتز هستند.در روی این غشاها رنگیزههای نوری یافت میشود.
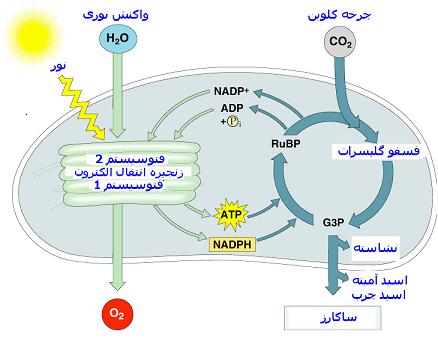
کلروپلاست جایگاه فتوسنتز
فتوسنتز فرایندی است که در گیاهان سبز برای تولید مواد غذایی بکار میرود که با استفاده از دیاکسید کربن و نور خورشید انجام میشود. فتوسنتز شامل دو سری واکنش وابسته به نور و غیر وابسته به نور است. واکنشهای غیر وابسته به نور یا واکنشهای تاریکی در استرومای کلروپلاست صورت میگیرد و طی آن انرژی شیمیایی لازم برای انجام واکنشهای مرحله نوری تامین میشود. این مرحله در بیشتر گیاهان در شب انجام میشود. در واکنشهای مرحله نوری با استفاده از دیاکسید کربن و نور خورشید انواع مختلف کربوهیدراتها ساخته میشود.
ژنوم کلروپلاست
کلروپلاست مانند میتوکندری DNA دارد و در آن همانند سازی ، رونویسی و پروتئین سازی مستقل از هسته صورت میگیرد. این فرایندها در بستره کلروپلاست انجام میگیرد. به نظر میرسد DNA کلروپلاستها مانند DNA میتوکندریها به غشای داخلی کلروپلاست چسبیدهاند. اندازه ژنوم کلروپلاست در تمام گیاهان مشابه است. DNA کلروپلاستها ملکولهایی حلقوی هستند. ژنوم کلروپلاست 120 ژن دارد و محصولات شناخته شده آنها شامل RNAهای ریبوزومی ، tRNAها ، برخی زیر واحدهای RNA پلیمراز ، برخی از پروتئینهای ریبوزومی و تعدادی از آنزیمهایی است که در فتوسنتز نقش دارند.
کلروپلاستزایی
کلروپلاست از تمایز پلاست اولیه و اتیوپلاست بوجود میآید. کلروپلاست مثل میتوکندری طی چرخه سلول بزرگ میشود و تقسیم دوتایی پیدا میکند. صفاتی که توسط DNA کلروپلاست تعیین میشوند، مانند وجود رنگدانههای عمل کننده در فتوسنتز در 3/2 گیاهای عالی از وراثت سیتوپلاسمی تبعیت میکنند و توارث اکثرا دو والدی میباشد. به عنوان مثال از آمیزش گیاه نر و مادهای که یکی کلروپلاست سالم و دیگری کلروپلاست معیوب دارد، گیاهانی حاصل میشوند که برگهای آنها دارای لکههای سبز و سفید هستند، لکههای سبز مربوط به کلروپلاست سالم است، در حالی که لکههای سفید مربوط به کلروپلاست معیوب هستند.
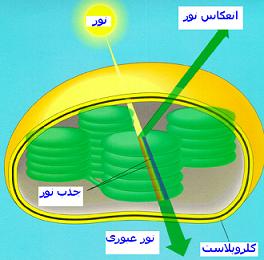
القای پلاست اولیه توسط نور و مراحل تمایز آن به کلروپلاست بالغ
- پلاست اولیه در سلولی که به تاریکی عادت دارد فقط غشای خارجی و داخلی دارد.
- در اثر مجاورت با نور ، کلروفیل ، فسفو لیپیدها ، بستره کلروپلاست و پروتئینهای تیلاکوئیدی ساخته میشوند و وزیکولهای کوچک از غشای داخلی جوانه میزنند.
- با بزرگ شدن پلاستها ، بعضی از وزیکولهای گرد ادغام میشوند و وزیکولهای پهن تیلاکوئیدی را تشکیل میدهند.
- در مراحل آخر تمایز کلروپلاست ، بعضی از وزیکولهای تیلاکوئیدی روی هم انباشته میشوند و گرانا (جمع گرانوم) را بوجود میآورند.
تکامل پلاستها از موجودات ابتدایی
از موجودات ابتدایی یا باکتریهای فتوسنتز کننده تکامل ساختارهای پلاستی در سه جهت انجام گرفته است.
- گسترش سطح نسبت به حجم که بخصوص برای کسب انرژی نورانی مناسب است.
- گزینش انواع مختلفی از رنگیزههای پذیرنده نور ، تشکیل گیرندههای نوری بسیار مختلف را امکان پذیر میسازد.
- تخصصی شدن اعمالی که منجر به تغییر ترکیب و ساختمان پلاست شده و موجب تولید انواع مختلف پلاستهای عمل کننده شده است که میتوانند به یکدیگر تبدیل شوند.
برگرفته از مجله و سایت رشد